The Larsson Quantum Lab
We develop and utilize tools for simulating and understanding quantum effects in molecular systems.

We develop and utilize tools for simulating and understanding quantum effects in molecular systems.
Our research focuses on developing and utilizing tools for simulating and understanding quantum effects in molecular systems. We are interested in both structural and dynamical properties of molecules. Our recent applications range from understanding entangled vibrational motion in floppy, protonated water clusters and using strong laser fields to control the motion of electrons to predicting complex bonding situations in strongly correlated transition-metal clusters.
Graduate students are welcome to apply either to the Chemistry Program or the Physics Program at UC Merced.
We are seeking two postdoctoral researchers to work on developing new real-time electron dynamics methods related to tensor network states and the density matrix renormalization group (DMRG).
Applicants with a background in chemistry/physics and with experience in method development and programming are welcome to apply. See the advertisement on the attochemistry project and the advertisement on the chiral molecular dynamics for further information and instructions on how to apply. For questions, please email Henrik Larsson at larsson [at] ucmerced .eδu.
Researchers interested in vibrational quantum dynamics and spectroscopy using tensor network states (DMRG, MCTDH, TTNS) are welcome to email Henrik Larsson for open postdoctoral positions.

To understand the dynamics of quantum many-body systems, it is essential to study excited eigenstates. While tensor network states have become a standard tool for computing ground states in computational many-body physics, obtaining accurate excited eigenstates remains a significant challenge. In this work, we develop an approach that combines the inexact Lanczos method, which is designed for efficient computation of excited states, with tree tensor network states (TTNSs). We demonstrate our approach by computing excited vibrational states for three challenging problems: (1) 122 states in two different energy intervals of acetonitrile (12-dimensional), (2) Fermi resonance states of the fluxional Zundel ion (15-dimensional),and (3) selected excited states of the fluxional and very correlated Eigen ion (33-dimensional). The proposed TTNS inexact Lanczos method is directly applicable to other quantum many-body systems.

Accurate vibrational spectra are essential for understanding how molecules behave, yet their computation remains challenging and benchmark data to reliably compare different methods are sparse. Here, we present high-accuracy eigenstate computations for the six-atom, 12-dimensional acetonitrile molecule, a prototypical, strongly coupled, anharmonic system. Using a density matrix renormalization group (DMRG) algorithm with a tree-tensor-network-state (TTNS) ansatz, a refinement using TTNSs as basis set, and reliable procedures to estimate energy errors, we compute up to 5,000 vibrational states with error estimates below 0.0007 cm-1. Our analysis reveals that previous works underestimated the energy error by up to two orders of magnitude. Our data serve as a benchmark for future vibrational spectroscopy methods and our new method offers a path toward similarly precise computations of large, complex molecular systems.

Compared to ground state electronic structure optimizations, accurate simulations of molecular real-time electron dynamics are usually much more difficult to perform. To simulate electron dynamics, the time-dependent density matrix renormalization group (TDDMRG) has been shown to offer an attractive compromise between accuracy and cost. However, many simulation parameters significantly affect the quality and efficiency of a TDDMRG simulation. So far, it is unclear whether common wisdom from ground state DMRG carries over to the TDDMRG, and a guideline on how to choose these parameters is missing. Here, in order to establish such a guideline, we investigate the convergence behavior of the main TDDMRG simulation parameters, such as time integrator, the choice of orbitals, and the choice of matrix-product-state representation for complex-valued non-singlet states. In addition, we propose a method to select orbitals that are tailored to optimize the dynamics. Lastly, we showcase the TDDMRG by applying it to charge migration ionization dynamics in furfural, where we reveal a rapid conversion from an ionized state with a σ character to one with a π character within less than a femtosecond.

The nitrate radical NO3 plays an important role in atmospheric chemistry, yet many aspects of its coupled and anharmonic vibronic structure remain elusive. Here, using an accurate, coupled full-dimensional diabatic potential that includes five electronic states, we revisit the vibronic spectrum associated with the electronic X~2A′2 state. Using recently developed tensor network state methods, we are able to compute more than 2500 vibronic states, thereby increasing the number of computed full-dimensional states by a factor of 50, compared to previous work. While we obtain good agreement with experiment for most of the assigned vibronic levels, for several others, we observe striking disagreement. Further, for the antisymmetric bending motion we find remarkably large symmetry-induced level splittings that are larger than the zero-order reference. We discuss non-negligible nonadiabatic effects and show that the Born-Oppenheimer approximation leads to significant errors in the spectrum.
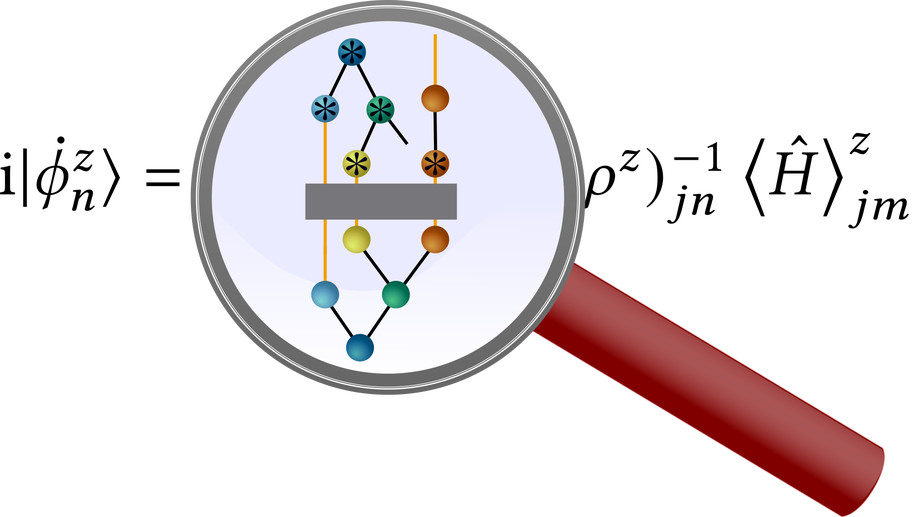
The multilayer multiconfiguration time-dependent Hartree (ML-MCTDH) method and the density matrix renormalization group (DMRG) are powerful workhorses applied mostly in different scientific fields. Although both methods are based on tensor network states, very different mathematical languages are used for describing them. This severely limits knowledge transfer and sometimes leads to re-inventions of ideas well known in the other field. Here, we review ML-MCTDH and DMRG theory using both MCTDH expressions and tensor network diagrams. We derive the ML-MCTDH equations of motions using diagrams and compare them with time-dependent and time-independent DMRG algorithms. We further review two selected recent advancements. The first advancement is related to optimizing unoccupied single-particle functions in MCTDH, which corresponds to subspace enrichment in the DMRG. The second one is related to finding optimal tree structures and on highlighting similarities and differences of MCTDH and DMRG theories. We hope that this contribution will foster more fruitful cross-fertilization of ideas between ML-MCTDH and DMRG.
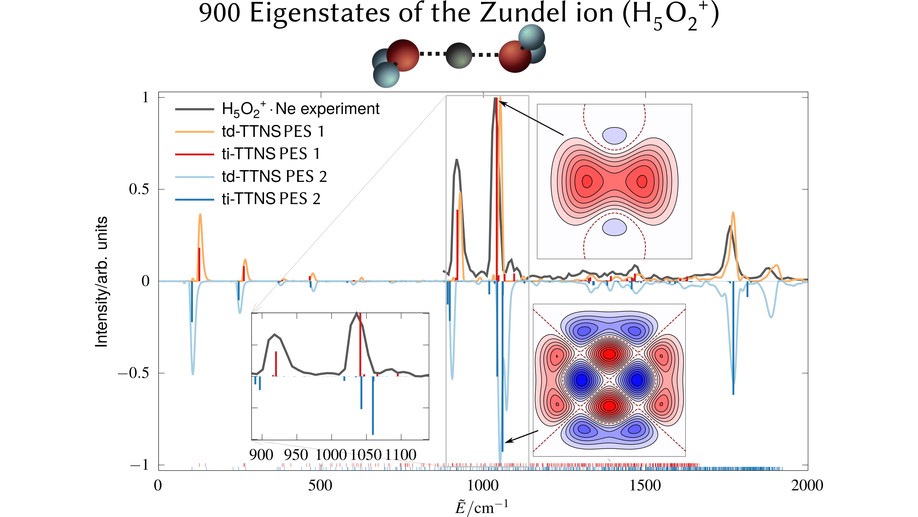
The infrared (IR) spectra of protonated water clusters encode precise information on the dynamics and structure of the hydrated proton. However, the strong anharmonic coupling and quantum effects of these elusive species remain puzzling up to the present day. Here, we report unequivocal evidence that the interplay between the proton transfer and the water wagging motions in the protonated water dimer (Zundel ion) giving rise to the characteristic doublet peak is both more complex and more sensitive to subtle energetic changes than previously thought. In particular, hitherto overlooked low- intensity satellite peaks in the experimental spectrum are now unveiled and mechanistically assigned. Our findings rely on the comparison of IR spectra obtained using two highly accurate potential energy surfaces (PESs) in conjunction with highly accurate state-resolved quantum simulations. We demonstrate that these high-accuracy simulations are important for providing definite assignments of the complex IR signals of fluxional molecules.
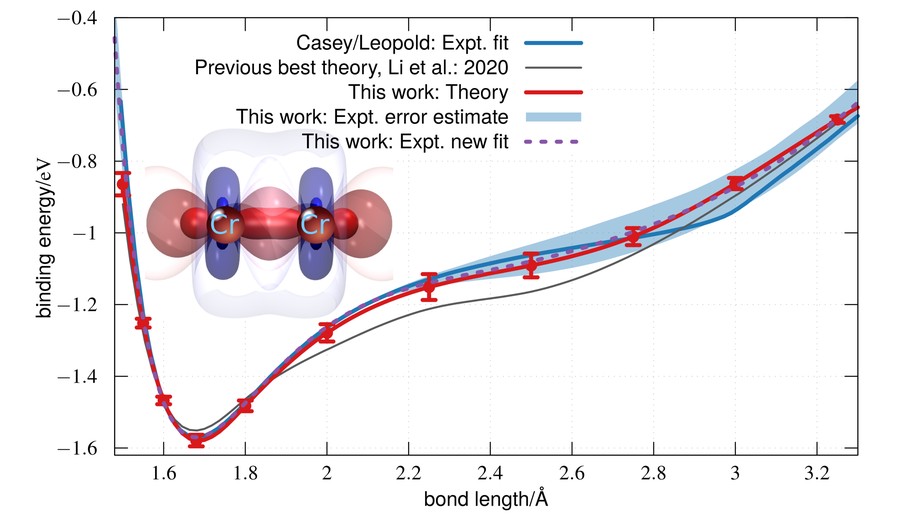
The complex electronic structure and unusual potential energy curve of the chromium dimer have fascinated scientists for decades, with agreement between theory and experiment so far elusive. Here, we present a new ab initio simulation of the potential energy curve and vibrational spectrum that significantly improves on all earlier estimates. Our data support a shift in earlier experimental assignments of a cluster of vibrational frequencies by one quantum number. The new vibrational assignment yields an experimentally derived potential energy curve in quantitative agreement with theory across all bond lengths and across all measured frequencies. By solving this long-standing problem, our results raise the possibility of quantitative quantum chemical modeling of transition metal clusters with spectroscopic accuracy.
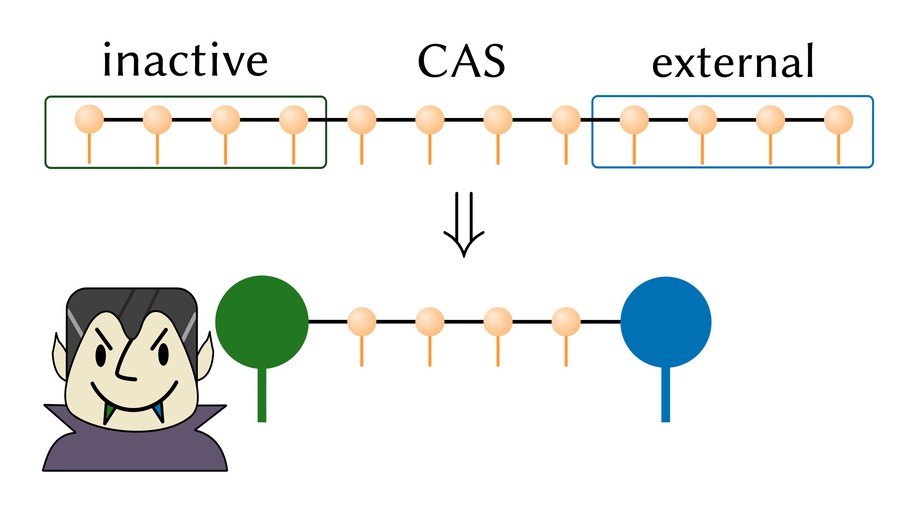
We explore various ways to group orbitals into clusters in a matrix product state (MPS). We explain how a generic cluster MPS can often lead to an increase in computational cost and instead propose a special cluster structure, involving only the first and last orbitals/sites, with a wider scope for computational advantage. This structure is a natural formalism to describe correlated multireference (MR) theories. We demonstrate the flexibility and usefulness of this approach by implementing various uncontracted MR configuration interaction, perturbation and linearized coupled cluster theories using an MPS with large cluster sites. Applications to the nitrogen dimer, the chromium dimer, and benzene, including up to triple excitations in the external space, demonstrate the utility of an MPS with up to two large sites. We use our results to analyze the quality of different multireference approximations.
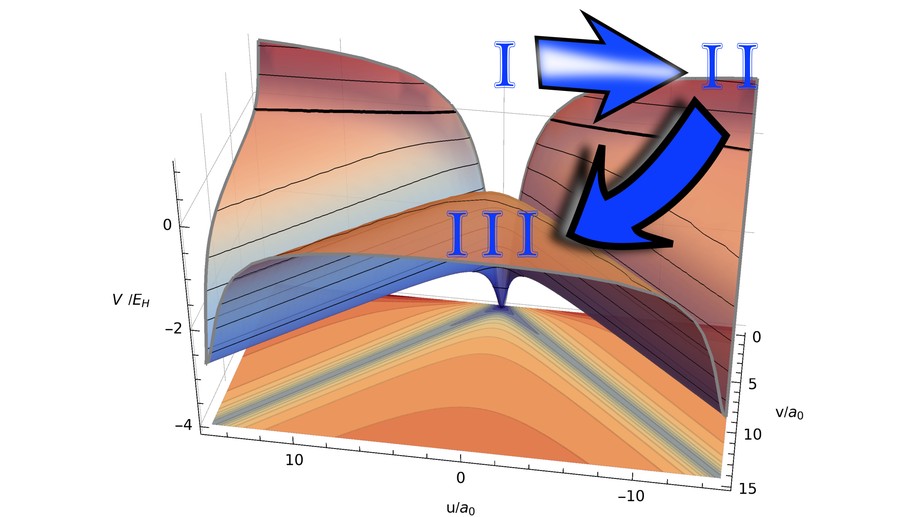
Our article sheds light on the control of the motion of the ionized electrons during double ionization. To first order, the field only drives the electron into the same direction (front-to-back motion). In this paper, our goal was to achieve the opposite, back-to-back motion, where both electrons are ejected into opposite directions. This is only possible via an interplay between the inter-particle forces and the potential. By extensive usage of four different control procedures (local control, derivative-free optimizations of basis expansions, Krotov method, and control of the classical equations of motions), we could identify pulse characteristics that lead to the desired back-to-back motion. Additionally, we were able to identify a simple, classical two-step mechanism. Besides these new physical results, this contribution also discusses new methodologies for using local control procedures for this difficult system and employs a new method to efficiently propagate the wavefunction in time [H. R. Larsson, B. Hartke and D. J. Tannor, J. Chem. Phys., 145, 204108 (2016)].
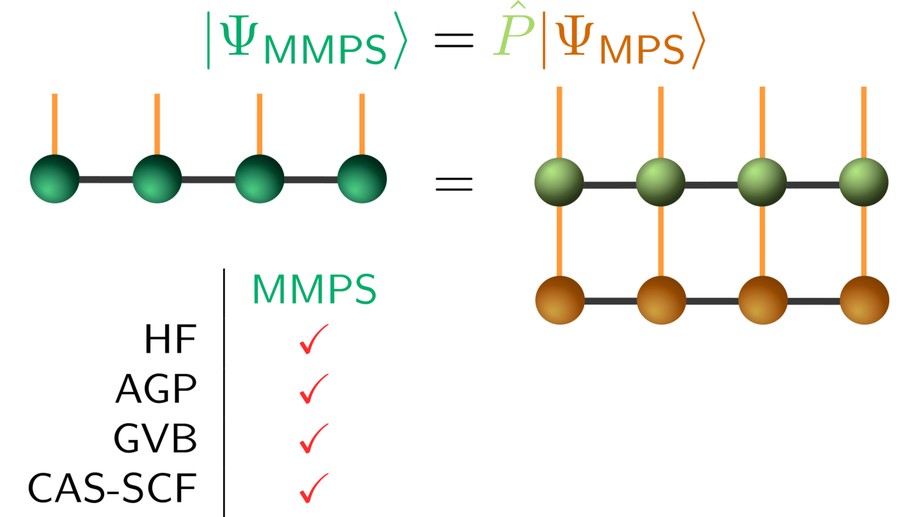
During my postdoctoral studies in the group of Prof. G. Chan (Caltech), I explored new ways to apply the density matrix renormalization group (DMRG) to bigger systems. In this article we explore a class of simple, qualitative wavefunctions, the minimal matrix product state (MMPS), which combines projection operators and the DMRG in order to yield a low-scaling method for quick explorations of the potential energy landscape of strongly correlated systems. Without jeopardizing low computational cost, MMPSs generalize well-established methods such as the projected Hartree-Fock-Bogoliubov (HFB)/antisymmetrized geminal power (AGP) method and the generalized valence bond (GVB) method. For the systems we studied, we show that MMPSs give correct qualitative behavior across the whole PES, often significantly improving aforementioned ansätze.
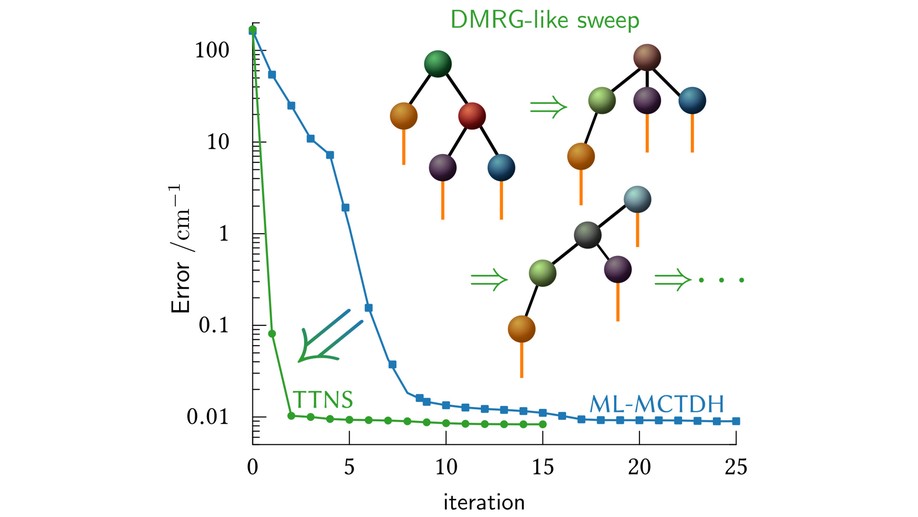
During my postdoctoral studies in the group of Prof. G. Chan (Caltech), as side project, I worked on the computation of vibrational spectra using tree-tensor network states (TTNS). There, I applied methods developed in the condensed matter community to molecular systems. Compared to established approaches for computing vibrational spectra, such as the multilayer multiconfiguration time-dependent Hartree (ML-MCTDH) method, the new approach is faster and much more robust and the diagrammatic language from condensed matter physics I use gains much more insights. I am currently working on using the TTNS method to compute high-lying vibrational states of the Zundel ion.
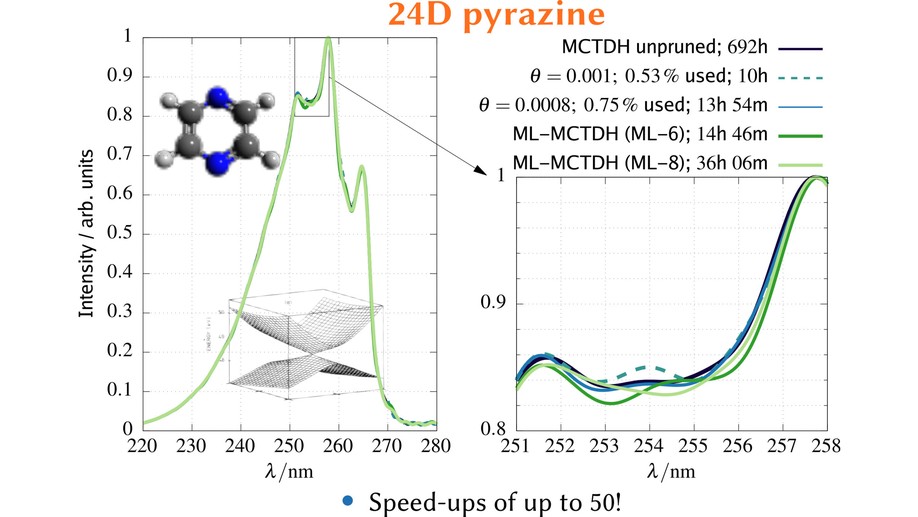
The main project of my PhD studies (in the groups of Prof. B. Hartke and Prof. D. Tannor) was the exploitation of sparsity in vibrational and electronic wavefunctions. Exploiting the sparsity means pruning of the underlying basis. This is similar to selected CI techniques for electronic structure calculations. Particularly successful was the application of dynamical pruning to the multiconfiguration time-dependent Hartree (MCTDH) method. While simple static pruning has already been studied 20 years ago by Worth [1], the conclusions from this mainly was that even static pruning requires too much bookkeeping such that this approach has quickly been abandoned. Due to this experience, many MCTDH developers advised me against developing methods based on pruning. Instead, I started working on a completely new MCTDH implementation with pruning. This was successful and the developed methods were applied to up to 24-dimensional systems and were up to 50 times faster than conventional MCTDH and competitive with state-of-the-art but more complicated multilayer MCTDH. The reasons for this speedup are newly developed algorithms, an efficient implementation, and a dynamical pruning instead of a simple static one. Furthermore, I applied, also for the first time, dynamical pruning both to the orbitals and the underlying grid representation. This means that the used basis changes at each time step at two different levels.
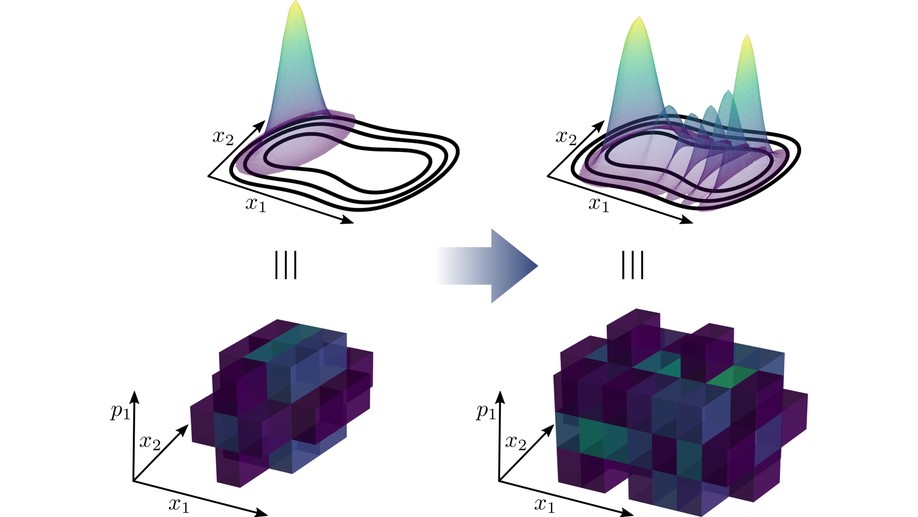
The main project of my PhD studies (in the groups of Prof. B. Hartke and Prof. D. Tannor) was the exploitation of sparsity in vibrational and electronic wavefunctions. Exploiting the sparsity means pruning of the underlying basis. This is similar to selected CI techniques for electronic structure calculations. However, here, pruning was used for solving the time-dependent Schrödinger equation. Since the wavefunction changes with time, also the sparsity pattern changes such that the pruning has to be done dynamically. In this publication, for the first time, I could show that pruning actually is faster than conventional methods. This was only possible by developing new algorithms adapted to the pruned basis and by identifying certain requirements the employed basis has to fulfill, most importantly orthogonality, in order to achieve an efficient algorithm. The developed algorithms deal with the update of the sparsity pattern and with the computation of the action of an operator onto a wavefunction. This can be understood as a transformation of sparse, high-dimensional tensors. Parts of the algorithms I developed are now used in the group of Prof. T. Carrington (Queen’s University) [1], who is one of the lead-developers of methods based on pruning.
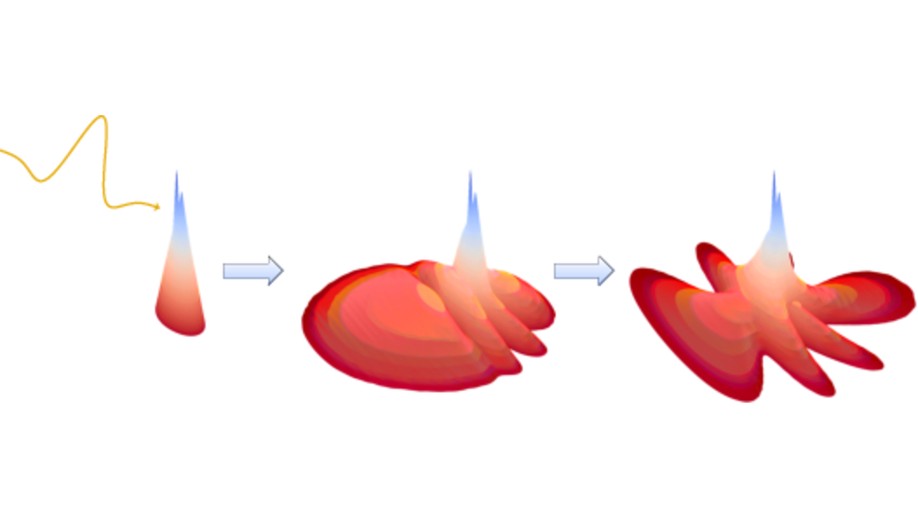
During my Master’s studies (in the group of Prof. M. Bonitz, Kiel University), I performed electron dynamics simulations utilizing a time-dependent multireference configuration interaction (TD-MR-CI) method. This method reduces the computational effort by considering only a selection of relevant configurations needed to describe the wavefunction, based on physical insights into the problem. With the help of a well-chosen coordinate system and a careful optimization of tensor transformations appearing in this method (more than 50,000 basis functions can be used), I could improve the methodology such that I was able to unravel correlation effects that occur during ionization dynamics of LiH in strong and short laser fields. In particular, I could show that, for intermediate field intensities, qualitatively wrong results are predicted if methods that are too simple are employed that take no correlation into account. My code is now used in the group of Prof. L. Madsen (Aarhus University) [1] [2].
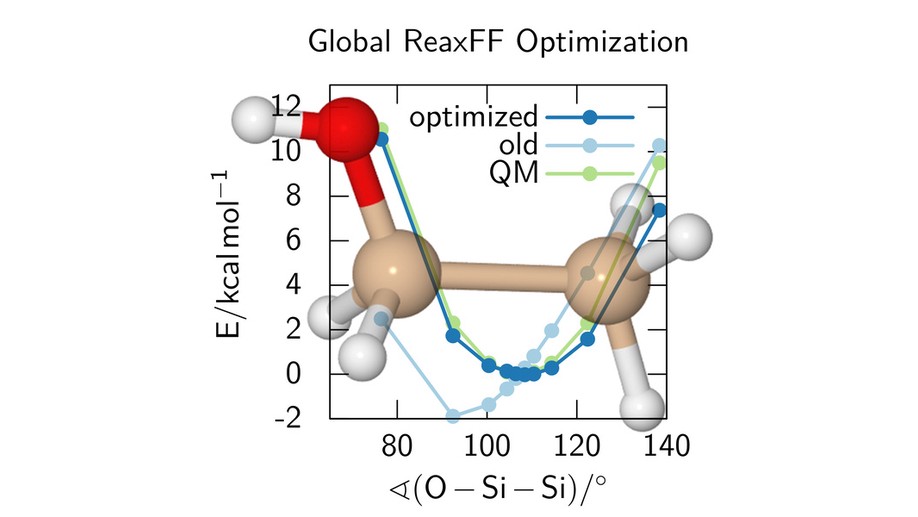
In my Bachelor’s studies (in the group of Prof. B. Hartke, Kiel University), I performed global optimization of the reactive force field ReaxFF. The development of force fields that are able to describe any chemical reactions is a highly complex task and the optimization of the parameters of the force fields is very demanding, due to the sheer number of parameters (up to 300), their strong and partially non-obvious interdependencies, and the complicated, highly nonlinear and rugged function that needs to be globally optimized. Analytic derivatives with respect to the parameters are typically not available. Despite these challenges, I successfully performed global optimization of ReaxFF for systems containing containing Si, O and H. My optimized force field outperformed that of the lead developer of ReaxFF.




